
所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。
大脑多畸形
該文的醫學專家

最近審查:04.07.2025
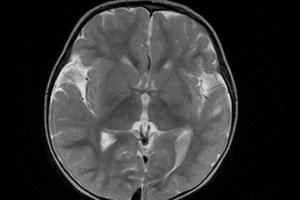
脑多小脑回畸形(源自拉丁语“gyrus”,意为“回旋”)是一种先天性缺陷,即大脑皮层细胞结构发生普遍改变,形成多个异常小的脑回。[ 1 ]
流行病學
据统计,各类脑发育不全中约有三分之一的病例存在脑皮质的先天性异常,但尚无关于孤立性多小脑回畸形患病率的数据。
原因 多雌雄同体
虽然多小脑回畸形的具体病因仍在研究中,但其病因的本质——与所有脑发育缺陷一样——在于胚胎发育过程中的偏差。[ 2 ]
在这种情况下,胎儿脑回形成过程会受到干扰——大脑皮层特征性褶皱的形成大约始于妊娠中期。这些褶皱的顶部形成脑回,而褶皱之间的凹陷则形成脑沟。在颅骨空间有限的情况下,脑回和脑沟的形成可以增加大脑皮层的面积。[ 3 ]
大脑皮层宫内发育障碍大多是由染色体异常和基因突变引起的。这可能是一个基因的突变,也可能是几个相邻基因的缺失。[ 4 ]
多小脑回畸形可以是孤立的,但也可与其他脑部异常同时发生 - 遗传性综合症,特别是 DiGeorge 综合症(22q11.2 染色体缺失综合症);[ 5 ] Adams-Oliver、Zellweger、Walker-Warburg 综合症;Aicardi 综合症(脑胼胝体发育不全)、Smith-Kingsmore 综合症(大头畸形)、Goldberg-Shprintzen 综合症(小头畸形和面部畸形)等。[ 6 ],[ 7 ]
風險因素
专家认为,多小脑回畸形的发生有以下风险因素:
- 遗传性基因缺陷;
- 胚胎中自发的基因突变;
- 毒素或感染对胎儿产生负面影响,主要是怀孕期间的巨细胞病毒感染;
- 胎盘灌注不足、胎儿缺氧导致的脑缺血;
- 各种原因的胎儿硬膜下出血。[ 8 ]
發病
尽管脑回形成的生理机制至今仍不清楚(目前存在多种解释),但多小脑回畸形的发病机制与脑结构神经发生的障碍有关,包括神经嵴胚胎细胞(神经母细胞)的迁移、分裂和增殖。此外,它还与上文提到的胎儿脑回形成障碍有关。
这些疾病会导致脑结缔组织膜(软脑膜)和蛛网膜(蛛网膜)出现缺陷,包括膜厚度和数量的变化、相邻脑回分子层的融合、膜血管化增加,并导致脑灌注受损(可能出现软皮质局部出血、皮层下白质水肿以及部分皮质萎缩)。[ 9 ]
在大脑皮层的组织发生中,其软壳基底膜起着重要作用。研究表明,多小脑回畸形和其他皮质缺陷可能与该膜的生长不稳定有关,其蛋白质和糖蛋白成分(IV型胶原、纤连蛋白、层粘连蛋白等)存在缺陷,从而导致皮层细胞结构的病理改变。
例如,在已发现的多小脑回畸形的基因突变中,值得关注的是位于16q21染色体上的GPR56(或ADGRG1)基因,该基因编码细胞粘附受体的膜G蛋白——细胞间接触调控胚胎形态发生过程,并决定组织形成的形式。该基因突变与双侧额顶叶多小脑回畸形的发生有关。[ 10 ]
症狀 多雌雄同体
如果多小脑回畸形影响儿童大脑的一侧,则称为单侧畸形;如果双侧大脑半球的皮质均受影响,则称为双侧畸形。多小脑回畸形主要影响背外侧皮质。
最初的迹象和随着时间的推移而出现的临床表现完全取决于大脑中哪些特定区域受到异常的影响。
单侧局灶性多小脑回畸形影响脑内相对较小的区域,最常扩散至额叶或额顶叶皮质,以及靠近侧裂沟的周围皮质。其表现为癫痫发作,可能不伴有其他神经系统症状。
双侧多小脑回畸形的表现包括反复癫痫发作、发育迟缓、肌肉无力、斜视(斗鸡眼)、吞咽困难和言语困难。
因此,除了频繁癫痫发作外,双侧额叶多小脑回畸形还表现为儿童一般和智力发育迟缓、痉挛性四肢瘫痪(下肢和上肢弛缓性瘫痪)、共济失调(运动协调障碍)、行走障碍(步态障碍),以及经常出现的共济失调(完全无法站立)和行走困难(无法行走)。
额顶叶多小脑回畸形或双侧额顶叶多小脑回畸形的特征性症状包括发育迟缓、认知障碍(中度或重度)、癫痫、凝视共济失调和斜视、共济失调、肌张力亢进。[ 11 ]
如果存在双侧外侧裂周围多小脑回畸形,那么在症状(出生时、婴儿期或接近两三岁时出现)中,最常见的是:四肢抽搐和痉挛、吞咽困难和流涎、面部、舌头、下颌和喉部肌肉的部分双侧麻痹,以及发育迟缓——一般和认知。
最严重的类型是双侧全身性多小脑回畸形,会影响整个大脑。这种疾病会导致严重的认知障碍、运动障碍和癫痫发作——持续性的强直阵挛性癫痫发作,难以或无法用药物控制。[ 12 ]
診斷 多雌雄同体
鑑別診斷
该病需与其他先天性脑异常进行鉴别诊断,包括巨脑回畸形、脑裂畸形、脑功能综合征以及儿童特发性全身性和局灶性癫痫。[ 14 ]
治療 多雌雄同体
預防
鉴于导致大脑皮层畸形发展的自发基因突变比例很大,因此预防是不可能的。
預測
多小脑回畸形在大多数情况下预后不良:87%-94%的患者会患上几乎无法治愈的癫痫,并伴有反复发作的癫痫。许多患有双侧畸形或一侧脑回一半以上受损的儿童在幼儿期死亡。