
所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。
儿童神经母细胞瘤:病因、诊断和治疗
該文的醫學專家

最近審查:04.07.2025
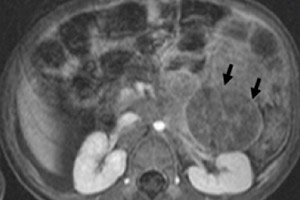
在儿科肿瘤学中,最常见的颅外肿瘤之一是儿童神经母细胞瘤,它是神经嵴神经母细胞(即交感神经系统的胚胎(未成熟)神经细胞)的胚胎恶性肿瘤。
流行病學
根据国际神经母细胞瘤风险组(INRG)的统计,神经母细胞瘤约占全球儿童所有肿瘤疾病的8%,发病率位居白血病和脑瘤之后,位居第三。
根据其他数据,神经母细胞瘤约占婴儿所有癌症的28%。超过三分之一的神经母细胞瘤病例诊断于一岁以下儿童;平均诊断年龄为19-22个月。超过90%的确诊病例发生在两至五岁儿童中(以男孩为主);发病高峰出现在两至三岁儿童中,五岁以上儿童的病例占比不到10%。
原因 神经母细胞瘤
在研究神经母细胞瘤的病因时,研究人员得出结论,这种儿童肿瘤是由于胚胎发生或出生后早期发育过程中偶尔发生的基因突变而发生的。但由于尚未发现致畸环境因素的影响,因此导致这些基因变异的原因尚不清楚。
这些肿瘤可发生在任何部位,包括纵隔、颈部、腹部、肾上腺、肾脏、脊柱和骨盆。
在极少数情况下,婴儿神经母细胞瘤可能与遗传突变有关。具体而言,位于2号染色体上的膜蛋白CD246基因突变——酪氨酸激酶ALK,该酶负责确保细胞间通讯,并在神经系统功能中发挥重要作用;以及位于4号染色体上的蛋白质PHOX2B基因突变,该基因参与神经细胞的成熟。
神经母细胞瘤也可能与儿童1 型神经纤维瘤病、贝克威斯-维德曼综合征和高胰岛素血症性低血糖症(神经母细胞瘤性胰腺炎)有关。
風險因素
如今,遗传已被公认为儿童罹患神经母细胞瘤的风险因素——家族史中存在此类肿瘤,以及宫内发育过程中与基因突变相关的先天性异常。对于在不同器官中同时出现多种肿瘤的情况,遗传风险尤其高。
研究人员尚未发现任何增加该肿瘤风险的外源因素。
發病
神经母细胞瘤的发生机制是由于神经嵴细胞(一种源自人类胚胎外胚层、在神经管边缘形成的双侧细胞系)的分化和成熟障碍所致。这些细胞能够迁移并分化成多种类型的细胞:感觉神经元和自主神经元、神经内分泌细胞、肾上腺髓质细胞、颅面软骨细胞和骨骼细胞,以及色素细胞。
在神经母细胞瘤中,迁移的神经母细胞不会成熟,而是继续生长和分裂,形成肿瘤。其形成的发病机制与以下基因突变有关:
- 胚胎神经嵴细胞中编码RBTN1蛋白的11号染色体上的LMO1基因的部分染色体序列或片段重复;
- 1q21.1染色体上NBPF10基因拷贝数发生改变,该基因编码控制人类神经干细胞增殖的DUF1220蛋白。这些疾病导致该染色体重复或缺失——部分DNA缺失;
- 随着肿瘤抑制基因 ATRX(位于染色体 Xq21.1 上)的变化;
- 由于2号染色体上N-Myc转录因子基因存在额外拷贝(扩增),该基因编码一种转录因子(DNA结合蛋白),调节其他基因的活性,并在胎儿组织和器官形成所需的蛋白质形成过程中控制前体细胞的增殖。该基因的扩增使其成为致癌基因,导致细胞周期紊乱、细胞增殖增加和肿瘤形成。
症狀 神经母细胞瘤
神经母细胞瘤的最初症状并不具有特异性,可能包括食欲不振(和体重减轻)、进食时疲劳、发烧和关节痛。
临床症状取决于原发肿瘤的位置和转移的存在(60-73%的病例会发生转移)。
原发性神经母细胞瘤通常位于肾上腺髓质,其起源与神经细胞相似。在一岁以下儿童中,35%-40% 的病例被诊断患有肾上腺神经母细胞瘤。其症状包括腹痛、发热、体重减轻、骨痛、贫血或伴随佩珀综合征:弥漫性肝损伤,伴有严重肝肿大和呼吸窘迫综合征。
腹膜后神经母细胞瘤或儿童腹膜后神经母细胞瘤随着生长,开始压迫膀胱或肠道,从而引起排尿或排便困难、腿部肿胀(男孩的阴囊肿胀)。
儿童纵隔神经母细胞瘤(纵隔神经母细胞瘤)常压迫上腔静脉,导致面部、颈部、手臂和上胸部肿胀(皮肤呈青红色,并伴有皮下结节)。患者会出现咳嗽、喘息、呼吸困难(气短)或吞咽困难;颈部、锁骨上方和腋窝淋巴结肿大。
肿瘤细胞扩散至骨髓,引起贫血、血小板减少、白细胞减少,并有出血倾向。
如果肿瘤转移到眶周,眼周会出现黑眼圈或淤青。这种肿瘤还会引起头痛、头晕、眼球突出(眼球凸出),以及由于神经末梢受压而导致的眼睑下垂(眼睑下垂)和瞳孔缩小(瞳孔缩小)。
儿童腹部神经母细胞瘤或腹腔神经母细胞瘤会导致腹部形成明显的封闭感、腹胀、食欲不振、便秘和血压升高。肿瘤压迫脊髓或神经根会导致肢体麻木无力,无法站立、爬行或行走。如果骨骼受到影响,可能会出现骨痛。
腹腔内3-4期肿瘤合并淋巴结损害时,肿瘤细胞可进入肾实质,形成儿童肾脏大面积神经母细胞瘤,导致肾脏功能障碍。
階段
- 第一阶段神经母细胞瘤是局限于身体某个部位的原发性肿瘤;两侧淋巴结不受影响。
- 神经母细胞瘤2期。2A期原发肿瘤局限于一个区域,但肿瘤较大;双侧淋巴结未受累。2B期肿瘤所在身体一侧的淋巴结转移阳性。
- 神经母细胞瘤3期:原发肿瘤跨越脊髓或身体中线,在淋巴结中发现单侧或双侧转移。
- 神经母细胞瘤4期:肿瘤已扩散至远处淋巴结、骨髓、骨骼、肝脏或其他器官。4S期:指一岁以下儿童,原发性肿瘤局限,并已扩散至皮肤、肝脏或骨髓。
国际神经母细胞瘤风险分期系统(INRGSS)
INRGSS 使用影像定义风险因素 (IDRF),即在影像学检查中发现的可能意味着肿瘤更难切除的因素。
INRGSS将神经母细胞瘤分为4个阶段:
- L1:肿瘤尚未从原发部位扩散,也尚未侵袭重要组织。肿瘤仅局限于身体某一部位,例如颈部、胸部或腹部。
- L2:肿瘤尚未扩散(转移)至远离其起始位置(例如,它可能已从腹部左侧生长至胸部左侧),但至少具有一个 IDRF。
- M:肿瘤已转移到身体的远处部位(MS阶段的肿瘤除外)。
- MS:18 个月以下儿童的转移性疾病,其中癌症仅扩散至皮肤、肝脏和/或骨髓。
並發症和後果
神经母细胞瘤的特点是并发症和后果,例如:
- 扩散(转移)至淋巴结、骨髓、肝脏、皮肤和骨骼;
- 脊髓压迫(可引起疼痛并导致瘫痪);
- 副肿瘤综合征的发展(由于肿瘤分泌的某些化学物质的作用,以及其细胞表达的抗原二唾液酸神经节苷脂GD2),表现为快速不自主的眼球运动,协调性受损,肌肉痉挛和腹泻;
- 完成主要治疗后复发(临床实践表明,高危神经母细胞瘤在 50% 的病例中复发)。
診斷 神经母细胞瘤
对儿童疑似神经母细胞瘤的诊断需要检查、实验室测试和影像学检查。
需进行血液和尿液检测,以检测儿茶酚胺(去甲肾上腺素和多巴胺)以及香草酸或香草扁桃酸(这些激素代谢过程中产生);血液检测神经特异性烯醇化酶,进行血清酶联免疫吸附试验 (ELISA),以及骨髓分析(通过穿刺抽取骨髓样本)。DNA 检测用于确定突变,并进行活检以对肿瘤组织进行细胞形态学检查。
活检样本采集后,会被送至实验室,由病理学家(接受过专门癌细胞识别培训的医生)在显微镜下进行观察。此外,通常还会对样本进行特殊的实验室检测,以确认肿瘤是否为神经母细胞瘤。
如果是神经母细胞瘤,实验室测试还可以帮助确定肿瘤生长或扩散的速度,以及哪种治疗方法最有效。
仪器诊断使用超声波、X 射线、MRI 或 CT、PET(引入 18F-氟脱氧葡萄糖)或 MIBG 扫描 - 使用间碘苄胍的闪烁显像来可视化肿瘤。 [ 1 ]
鑑別診斷
鉴别诊断包括良性神经节细胞瘤、神经节细胞母细胞瘤、横纹肌肉瘤、肾母细胞瘤。
治療 神经母细胞瘤
神经母细胞瘤的治疗取决于患者的风险组(肿瘤进展的阶段)、肿瘤的定位、肿瘤细胞的基因组特征以及患儿的年龄。治疗方案可能包括监测、手术、化疗、放射治疗、免疫治疗和造血干细胞移植。
儿童神经母细胞瘤的新辅助或辅助(术前或术后)化疗,与任何癌症化疗一样,采用疗程给药:连续给药数日,然后停药,让身体恢复。疗程通常每三至四周重复一次。
使用以下药物(及其组合):环磷酰胺、顺铂或卡铂、阿霉素(阿霉素)、长春新碱、依托泊苷。
化疗药物的常见副作用包括脱发、食欲不振、疲劳、恶心呕吐、口腔溃疡、腹泻或便秘。化疗会损害骨髓,导致血细胞计数下降。
靶向免疫疗法(针对肿瘤抗原GD2)使用单克隆抗体(抗GD2 MAb)Dinutuximab(Unituxin)和Naxitamab等药物。这些药物通过静脉输注,联合使用粒细胞-巨噬细胞集落刺激因子(细胞因子GM-CSF)和白细胞介素-2。
这些药物的副作用包括疼痛(通常非常严重)、血压降低、心率加快、呼吸急促(可能伴有气道肿胀)、体温升高、恶心、呕吐和腹泻、血液细胞和矿物质成分的变化。
为了降低大剂量化疗和干细胞移植后癌症复发的风险,患有高危神经母细胞瘤的儿童可以接受全身性类维生素 A 药物、13-顺式视黄酸(异维甲酸)治疗。[ 2 ]
神经母细胞瘤的手术治疗——肿瘤切除,例如开放性肾上腺切除术或腹腔镜肾上腺神经母细胞瘤切除术;淋巴切除术(切除受影响的淋巴结)等。[ 3 ]
对于高危神经母细胞瘤,可采用放射疗法。[ 4 ]
預防
鉴于儿童神经母细胞瘤的病因,唯一的预防措施可能是在计划怀孕时进行遗传咨询。但需要注意的是,这种肿瘤仅占1-2%的病例与遗传突变有关。
預測
婴儿神经母细胞瘤具有自发消退的能力。
预后标志物
- 高风险肿瘤以及所有年龄段和所有阶段(4S 期除外)儿童中的神经母细胞瘤 - N-MYC 基因表达增加和 N-Myc 致癌基因扩增 - 预后不良,会影响预期寿命。
- 肿瘤细胞缺失1号或11号染色体的某些部分(称为1p或11q缺失)会导致预后较差。17号染色体额外增加(17q增加)也会导致预后较差。
- 含有大量DNA的神经母细胞瘤细胞预后较好,尤其是2岁以下的儿童。
- 具有更多神经营养因子受体,特别是神经生长因子受体TrkA的神经母细胞瘤预后更好。
按儿童肿瘤组 (COG) 风险组划分的生存率
- 低危组:低危组儿童5年生存率大于95%。
- 中危组:中危组儿童的5年生存率为90%~95%。
- 高危人群:高危组儿童5年生存率约为50%。
约15%的儿童癌症死亡是由神经母细胞瘤引起的。这种高危恶性肿瘤的长期生存率不超过40%。总体五年生存率为67-74%,其中一至四岁年龄段的生存率为43%,出生后一岁内确诊的神经母细胞瘤生存率超过80%。