
所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。
儿童遗传性肾炎(阿尔波特综合征
該文的醫學專家

最近審查:05.07.2025
遗传性肾炎(Alport综合征)是一种基因决定的遗传性非免疫性肾小球疾病,表现为血尿(有时伴有蛋白尿),肾功能进行性减退,并发展为慢性肾衰竭,常合并神经性耳聋和视力障碍。
这种疾病最早由 L.G. Guthrie 于 1902 年描述,他观察到一个家族中几代人都患有血尿。1915 年,AF Hurst 描述了同一家族成员患上尿毒症的情况。1927 年,A. Alport 首次在几位患有血尿的亲属中发现听力损失。20 世纪 50 年代,描述了类似疾病的眼部病变。1972 年,在患有遗传性血尿的患者中,在肾脏组织形态学研究中,Hinglais 等人发现肾小球基底膜扩张不均匀且分层。1985 年,遗传性肾炎的遗传基础被确定——IV 型胶原基因突变(Fiengold 等人,1985 年)。
通过对该疾病遗传特性的研究,我们得出结论:遗传性肾炎(伴或不伴听力损失)的表型表现差异是由于突变基因的表达程度不同所致。因此,目前所有临床变异型均被视为同一种疾病的表现,“遗传性肾炎”一词与“Alport综合征”同义。
据流行病学研究,遗传性肾炎的发病率为每十万名儿童中有17人。
 [
[ 阿尔波特综合征的病因
该疾病的遗传基础是IV型胶原蛋白a-5链基因突变。该类型胶原蛋白普遍存在于肾脏基底膜、耳蜗、晶状体囊、视网膜和角膜中,这已在使用针对该胶原蛋白片段的单克隆抗体进行的研究得到证实。近期,已有研究指出,使用DNA探针进行遗传性肾炎的产前诊断具有可行性。
强调对所有家庭成员进行DNA探针检测以识别突变基因携带者的重要性,这对于为患有该疾病的家族进行医学和遗传咨询至关重要。然而,高达20%的家族中没有患肾病的亲属,这表明异常基因的自发突变频率很高。大多数遗传性肾炎患者的家族中都有患有肾病、听力损失和视力障碍的亲属;有一个或多个祖先的近亲结婚很重要,因为在有亲属关系的婚姻中,从父母双方获得相同基因的可能性会增加。目前已确定的遗传途径包括常染色体显性遗传、常染色体隐性遗传以及显性X连锁遗传。
在儿童中,最常见的遗传性肾炎有三种类型:Alport 综合征、无听力损失的遗传性肾炎和家族性良性血尿。
Alport综合征是一种伴有听力障碍的遗传性肾炎。该病由肾脏、耳和眼结构的肾小球基底膜胶原蛋白结构共同缺陷引起。经典Alport综合征的基因位于X染色体长臂21-22q位点。在大多数情况下,该病以显性方式遗传,与X染色体相关。因此,Alport综合征在男性中更为严重,因为在女性中,突变基因的功能由第二条未受损染色体的健康等位基因补偿。
遗传性肾炎的遗传基础是IV型胶原α链基因突变。已知IV型胶原G有六条α链:α5和α6链(Col4A5和Col4A5)的基因位于X染色体长臂21-22q区;α3和α4链(Col4A3和Col4A4)的基因位于第2条染色体;α1和α2链(Col4A1和Col4A2)的基因位于第13条染色体。
大多数病例(80-85%)检测出该病的X连锁遗传模式,与Col4A5基因因缺失、点突变或剪接障碍而受损有关。目前已发现超过200种Col4A5基因突变,这些突变导致IV型胶原α5链合成中断。这种遗传类型的疾病在男女儿童中均有发病,但男孩的病情更为严重。
负责IV型胶原蛋白a3和a4链合成的Col4A3和Col4A4基因位点的突变是常染色体遗传的。研究表明,16%的遗传性肾炎病例为常染色体显性遗传,6%的患者为常染色体隐性遗传。目前已知的Col4A3和Col4A4基因突变类型约为10种。
突变导致IV型胶原蛋白的组装过程发生异常,进而导致其结构破坏。IV型胶原蛋白是肾小球基底膜、耳蜗和晶状体的主要成分之一,其病理改变可在遗传性肾炎的临床诊断中发现。
IV型胶原蛋白是肾小球基底膜的一部分,主要由两条a1链(IV)和一条a2链(IV)组成,此外还包含a3、a4和a5链。在X连锁遗传中,最常见的情况是Col4A5基因突变导致IV型胶原蛋白结构中a3、a4、a5和a6链缺失,而肾小球基底膜中a1和a2链的数量增加。该现象的机制尚不清楚,推测其原因是mRNA的转录后变化。
Alport综合征早期,由于肾小球基底膜IV型胶原结构中缺少a3、a4、a5链,导致基底膜变薄、脆性增加,临床表现以血尿(血尿伴蛋白尿或仅有蛋白尿较少)、听力下降、圆锥形晶状体等为多见。病情进一步进展,晚期基底膜增厚、通透性降低,V型和VI型胶原增生,表现为蛋白尿增多、肾功能减退。
遗传性肾炎的基因突变性质在很大程度上决定了其表型表现。如果X染色体缺失,同时伴有负责IV型胶原α5链和α6链合成的Col4A5和Col4A6基因突变,则会导致Alport综合征,并伴有食管和生殖器平滑肌瘤病。研究数据显示,与该基因的点突变相比,如果Col4A5基因突变伴有缺失,则病理过程更为严重,肾脏损害伴有肾外表现,并可能早期发展为慢性肾衰竭。
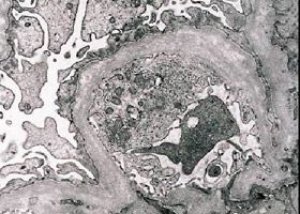
形态学上,电子显微镜检查可显示肾小球基底膜(尤其是致密板)变薄和分层,并可见电子致密颗粒。同一患者的肾小球病变可能呈现异质性,从轻微的局灶性系膜病变到肾小球硬化症均可表现。Alport综合征中的肾小球炎始终呈免疫阴性,这与肾小球肾炎相区别。其特征性表现包括肾小管萎缩、淋巴组织细胞浸润以及含有脂质包涵体的“泡沫细胞”(即噬脂细胞)。随着病情进展,肾小球基底膜会增厚并明显破坏。
免疫系统的某些变化有所显现。遗传性肾炎患者的血液中Ig A水平下降,而IgM浓度趋于升高;IgG水平在疾病早期可能升高,在晚期则下降。IgM和IgG浓度的升高或许是对IgA缺乏的一种代偿反应。
T淋巴细胞系统的功能活性降低;负责合成Ig A的B淋巴细胞选择性减少,免疫的吞噬联系被破坏,主要是由于中性粒细胞趋化性和细胞内消化过程的破坏
在对Alport综合征患者进行肾活检时,电子显微镜数据显示肾小球基底膜的超微结构变化:肾小球基底膜变薄、结构破坏、分裂,厚度改变且轮廓不均匀。在遗传性肾炎的早期阶段,这种缺陷决定了肾小球基底膜变薄且易碎。
肾小球膜变薄是一种较为有利的体征,在女孩中更为常见。遗传性肾炎中更常见的电子显微镜体征是基底膜分裂,其破坏程度与病变的严重程度相关。
儿童阿尔波特综合征的症状
Alport 综合征的首发症状为孤立性尿道综合征,最常在三岁以下儿童中发现。大多数情况下,该疾病是偶然发现的。尿道综合征是在儿童预防性体检中、入托儿所前或急性呼吸道感染 (ARVI) 期间发现的。如果在 ARVI 期间尿液中出现病理改变,则可确诊。遗传性肾炎与获得性肾小球肾炎不同,没有潜伏期。
在疾病的初期,儿童的健康几乎不受损害,其特征是尿路综合征的持续性和顽固性。主要症状之一是不同程度的血尿,在100%的病例中均可见。在呼吸道感染期间或之后、体力活动或预防性疫苗接种后,血尿程度会加重。蛋白尿在大多数情况下不超过1克/天,在疾病初期可能不稳定,随着病情进展,蛋白尿会增加。尿沉渣中可能周期性地出现以淋巴细胞为主的白细胞尿,这与间质改变的发生有关。
随后,部分肾功能受损,患者的一般状况恶化:出现中毒、肌肉无力、动脉低血压,通常出现听力障碍(尤其是男孩),有时还会出现视力障碍。中毒表现为苍白、疲劳和头痛。在疾病的早期阶段,听力损失在大多数情况下只能通过听力图检测出来。Alport 综合征的听力损失可能发生在儿童的不同时期,但最常见的听力损失是在 6-10 岁时诊断出来的。儿童听力损失从高频开始,在气导和骨传导中达到显著程度,从声传导听力损失发展到声感知听力损失。听力损失可能是该疾病的首发症状之一,并且可能先于尿路综合征出现。
20% 的 Alport 综合征患者会出现视觉器官异常。最常见的异常是晶状体异常:球形视、前圆锥晶状体、后圆锥晶状体或混合性晶状体,以及各种白内障。Alport 综合征家族中近视发病率很高。许多研究人员不断注意到这些家族的双侧黄斑周围改变,表现为黄体中出现亮白色或淡黄色颗粒。他们认为这种体征是 Alport 综合征的一个持续症状,具有很高的诊断价值。KS Chugh 等人(1993 年)在一项眼科研究中发现,66.7% 的 Alport 综合征患者视力下降,37.8% 出现前圆锥晶状体,22.2% 出现视网膜斑点,20% 出现白内障,6.7% 出现圆锥角膜。
一些患有遗传性肾炎的儿童,尤其是在出现肾衰竭时,会出现明显的身体发育迟缓。随着肾衰竭的进展,动脉高血压会逐渐出现。在儿童中,高血压更常见于青少年和老年人群体。
遗传性肾炎患者的特点是存在多种(超过5-7种)结缔组织畸形特征。患者最常见的结缔组织特征包括眼距过宽、腭高、咬合异常、耳廓形状异常、小指弯曲以及足部“凉鞋间隙”。遗传性肾炎的特点是,畸形特征在家族内具有一致性,并且在疾病遗传的先证者亲属中分布频率较高。
疾病早期,仅表现为部分肾功能下降:氨基酸转运、电解质、浓缩功能、酸生成等。后期,肾单位近端和远端的功能状态均受到影响,并出现合并性部分功能障碍。肾小球滤过率下降出现较晚,多见于青少年时期。随着遗传性肾炎的进展,会出现贫血。
因此,遗传性肾炎的特点是其病程呈阶段性:起初为潜伏期或临床症状隐匿,表现为尿路综合征的轻微改变;随后逐渐进入失代偿期,肾功能下降,出现明显的临床症状(中毒、乏力、发育迟缓、贫血)。临床症状的出现通常与炎症反应的分层无关。
遗传性肾炎可以在不同的年龄段表现出来,这取决于基因的作用,基因在一定时期内处于抑制状态。
Alport综合征的诊断
建议采用以下标准:
- 每个家庭中至少有两名肾病患者;
- 血尿为病因不明的肾病的主要症状;
- 至少一名家庭成员存在听力损失;
- 一个或多个亲属出现慢性肾衰竭。
在各种遗传性和先天性疾病的诊断中,综合检查方法占据重要地位,尤其要重视在整理儿童家谱时获得的数据。Alport综合征的诊断在患者身上发现以下4种典型体征中的3种即可:家族中有血尿和慢性肾衰竭病史;存在神经性听力损失;患者存在视力病变;在活检电子显微镜下发现肾小球基底膜裂解征象,厚度改变且轮廓不均匀。
患者的检查应包括临床和遗传学研究方法;有针对性的病史研究;结合诊断重要标准对患者进行一般检查。在代偿期,只有通过关注遗传负担、低血压、多种胚胎发育不良征兆、泌尿系统综合症改变等综合征才能发现病理。在失代偿期,可能出现肾外症状,例如严重中毒、乏力、身体发育迟缓、贫血,这些症状随着肾功能逐渐减退而出现和加剧。在大多数肾功能减退的患者中,会出现以下症状:酸合成和氨基酸合成减少;50% 的患者出现肾脏分泌功能显著下降;尿液光密度波动范围有限;滤过节律紊乱,随后肾小球滤过率降低。当患者血清尿素水平升高(超过0.35克/升)持续3-6个月或更长时间,并且肾小球滤过率降低至正常的25%时,即可诊断为慢性肾衰竭。
遗传性肾炎的鉴别诊断主要应与血尿型获得性肾小球肾炎进行。获得性肾小球肾炎多发于急性感染后2-3周,并伴有肾外体征,包括发病第一天即出现高血压(遗传性肾炎则表现为低血压),发病初期肾小球滤过率降低,部分肾小管功能不受影响,而遗传性肾小球肾炎则存在这些损害。获得性肾小球肾炎的血尿和蛋白尿更为明显,血沉增快。典型的肾小球基底膜改变是遗传性肾炎的特征,具有诊断价值。
代谢性肾病的鉴别诊断适用于慢性肾衰竭,以及家族性临床表现为异质性肾病的患者,肾病的病因可能涵盖从肾盂肾炎到尿路结石等一系列疾病。儿童常有腹痛,并伴有周期性排尿不适,尿沉渣中可检出草酸盐。
如果怀疑患有遗传性肾炎,应将患者转诊至专门的肾病科以明确诊断。
需要檢查什麼?
如何檢查?
需要什麼測試?
誰聯繫?
Alport综合征的治疗
该方案包括限制剧烈体力活动和呼吸新鲜空气。饮食均衡,富含足量的蛋白质、脂肪和碳水化合物,并兼顾肾功能。最重要的是发现和治疗慢性感染灶。使用以下药物:ATP、辅羧酶、吡哆醇(每日最高剂量50毫克)、氯化肉碱。疗程每年2-3次。对于血尿,医生会开草药——荨麻、黑果酸莓汁、西洋蓍草。
国内外文献中均有关于泼尼松龙及细胞抑制剂治疗的报道,但疗效难以判断。
对于慢性肾衰竭,可使用血液透析和肾移植。
遗传性肾炎目前尚无特效(有效的)治疗方法。所有治疗措施均旨在预防和减缓肾功能的衰退。
饮食应均衡,高热量,并考虑到肾脏的功能状态。在没有功能障碍的情况下,儿童的饮食应包含足够的蛋白质、脂肪和碳水化合物。如果出现肾功能障碍的体征,则应限制蛋白质、碳水化合物、钙和磷的摄入量,以延缓慢性肾衰竭的发展。
应限制体力活动;建议儿童避免运动。
应避免与传染病患者接触,降低患急性呼吸道疾病的风险。必须对慢性感染灶进行卫生处理。患有遗传性肾炎的儿童无需接种预防性疫苗,仅在出现流行病学指征时才可接种疫苗。
激素和免疫抑制疗法对遗传性肾炎无效。有迹象表明,长期使用环孢素A和血管紧张素转换酶(ACE)抑制剂可产生一些积极效果(减少蛋白尿并减缓病情进展)。
在治疗患者时,使用改善新陈代谢的药物:
- 吡哆醇-2-3毫克/千克/天,分3次服用,持续4周;
- 辅羧酶——隔日肌肉注射50毫克,共注射10-15次;
- ATP——每隔一天肌肉注射1毫升,注射10-15次;
- 维生素 A——1000 IU/年/天,1 次剂量,持续 2 周;
- 维生素 E — 1 毫克/千克/天,1 次剂量,持续 2 周。
这种疗法有助于改善患者的整体状况,减少肾小管功能障碍,每年进行3次疗程。
左旋咪唑可用作免疫调节剂 - 每周 2-3 次,每次 2 mg/kg/天,每次服用间隔 3-4 天。
据研究资料显示,高压氧对血尿严重程度及肾功能不全有积极作用。
治疗遗传性肾炎最有效的方法是及时进行肾移植。在这种情况下,移植肾不会复发;少数病例(约5%),移植肾可能会出现与肾小球基底膜抗原相关的肾炎。
一个有前景的方向是产前诊断和基因工程治疗。动物实验表明,将负责IV型胶原α链合成的正常基因高效地转移到肾脏组织中,之后可以观察到正常胶原结构的合成。