
所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。
碱丙酮尿症是一种先天性酶异常
該文的醫學專家

最近審查:04.07.2025
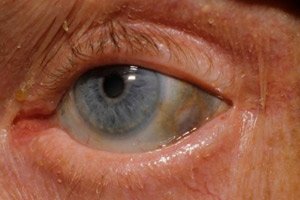
黑尿症是一种非常罕见的代谢紊乱疾病,是指氨基酸酪氨酸代谢的先天性异常。
该综合征也可称为尿黑酸氧化酶缺乏症、尿黑素尿症、遗传性褐黄病或黑尿病。[ 1 ]
流行病學
据统计,每100万人中,黑酸尿症的发病率不超过9例,而大多数欧洲国家,每10万至25万活产婴儿中,才会出现1例黑酸尿症。
在欧洲国家中,斯洛伐克(尤其是面积相对较小的西北部地区)是个例外,那里每19,000名新生儿中就有1例尿黑酸尿症。这很可能是因为居住在那里的斯洛伐克罗姆人家庭中,近亲结婚(堂兄弟间通婚)的比例是欧洲最高的:10-14%。[ 2 ]
原因 碱通尿症
黑尿症是一种先天性芳香族(同环)α-氨基酸酪氨酸分解代谢障碍(代谢分解障碍),其确切病因已得到明确:此类代谢障碍是由3号染色体上数千个基因之一的纯合突变或复合杂合突变引起的,更确切地说,是位于染色体长臂3q21-q23位点的HGD基因。该基因编码肝酶尿黑酸-1,2-双加氧酶的核苷酸序列 [ 3 ](也称为尿黑酸氧化酶或尿黑酸氧化酶),这是一种含铁金属蛋白,是体内酪氨酸分解的必要步骤之一。[ 4 ],[ 5 ]
因此,黑黑酸尿症是尿黑酸-1,2-双加氧酶缺陷所致,或者更准确地说,是基因决定的该酶缺陷或完全缺失的结果。 [ 6 ]
尿黑酸尿症是一种先天性酶缺乏症,属于常染色体隐性遗传,也就是说,如果孩子患有尿黑酸尿症,父母双方都必须携带这种酶的修饰基因,因为父母双方只会从两个基因拷贝中遗传给孩子一个。
据最新资料显示,HGD基因修饰有超过两百种变异,其中最常见的是错义突变、易位和剪接。
風險因素
罹患这种先天性酶病的唯一风险因素是家族史中存在该病,并且遗传了两个 HGD 基因的修饰副本,如果父母没有表现出尿黑酸尿症(遗传这种异常的风险为 25%),或者父母一方患有这种疾病。[ 7 ]
發病
酪氨酸在蛋白质的合成、色素蛋白(皮肤色素黑色素)的产生以及甲状腺激素和儿茶酚胺神经递质中起着关键作用。
细胞内酪氨酸含量的调节机制非常复杂,人体通过分解酪氨酸来使其含量过量恢复正常。酪氨酸的分解代谢过程与所有芳香族氨基酸一样,是多阶段的,并且包含多个阶段。酪氨酸代谢分解的每个阶段都伴随着特定酶的参与和中间化合物的形成。
因此,氨基酸首先被分解为对羟基苯丙酮酸,进而转化为苯并酮——2,5-二羟基苯乙酸或尿黑酸。然后,苯并酮应该转化为马来乙酸,但这并没有发生。[ 8 ]
而尿黑酸尿症的发病机制包括在尿黑酸形成阶段酪氨酸分解代谢的生化反应停止:根本没有分解尿黑酸所需的酶——尿黑酸氧化酶。
尿黑酸不被人体利用,会随着肾脏排泄而蓄积。此外,尿黑酸还会被氧化为苯醌乙酸酯(苯醌乙酸),苯醌乙酸酯与组织和体液分子结合,形成类似黑色素的生物高分子化合物。
这些中间产物在组织中积累,导致软骨组织胶原结构破坏,从而降低其弹性——出现尿黑酸尿症的许多临床症状并引发并发症。
症狀 碱通尿症
新生儿和婴儿的尿黑酸尿症的特征是尿液颜色变深。尿布、尿片和内衣上的尿液暴露在空气中时会变成深棕色;这是由于尿黑酸的积聚和释放,尿黑酸会被氧化成苯并喹乙酸酯。[ 9 ]
由于缺乏其他症状,幼儿的尿黑酸尿症通常无法及时发现,因为尿液在排尿数小时后可能会变暗。一些数据显示,在12个月以下出生时患有这种酶缺乏症的儿童中,只有五分之一在临床上被确诊。因此,父母重视对婴儿的护理至关重要。
此外,早期症状还包括眼巩膜和耳鼻软骨的色素沉着(蓝灰色),通常被称为褐黄病。[ 10 ]
随着时间的推移,还会出现其他症状:
- 颧骨、腋窝和生殖器皮肤色素沉着严重;
- 衣服与身体出汗的部位接触时会染色;
- 全身无力症发作;
- 声音嘶哑。
应该记住,如上所述,黑尿症和褐黄病是酪氨酸分解代谢同一种疾病的同义词。
枫糖尿症和尿黑酸尿症。先天性枫糖尿症或白蛋白血症也是一种代谢性疾病,具有相同的遗传模式,甚至突变发生在同一染色体上,但会影响编码支链α-酮酸脱氢酶酶复合物的基因。因此,人体无法分解某些蛋白质成分,特别是氨基酸亮氨酸、异亮氨酸和缬氨酸。患有这种疾病的人,尿液(和耳垢)会散发出甜味;此外,这种有机酸血症的临床表现包括色素减退、血压波动、癫痫发作、呕吐和腹泻、血糖水平下降、酮症酸中毒、幻觉等。儿童患者的死亡率相当高;成人患者如果不接受治疗,可能会因脑水肿而昏迷甚至死亡。
白化病和黑黑尿症仅因酪氨酸而“联系在一起”。白化病(包括眼皮肤白化病)是由影响黑色素生成的基因突变引起的。11号染色体(11q14.3)上的TYR基因存在先天性变异,该基因编码酪氨酸酶,这是一种含铜的黑素体酶,是酪氨酸代谢产物形成皮肤色素所必需的。这种疾病比黑黑尿症更常见。
鑑別診斷
鉴别诊断包括新生儿血色素沉着症和急性肝功能衰竭、黑素尿、急性间歇性卟啉症、噬血细胞性淋巴组织细胞增生症、原发性线粒体病理、类风湿性关节炎、强直性脊柱炎。
誰聯繫?
治療 碱通尿症
尿黑酸尿症的主要治疗方法是口服大剂量(每日至少1000毫克)抗坏血酸。对于儿童,这会增加尿液中尿黑酸的排泄;对于成人,这会增加尿液中其衍生物苯醌乙酸的含量,并减缓其与关节结缔组织结构和胶原蛋白的结合。[ 16 ]
西欧的诊所正在测试一种名为尼替西农(Orfalin)的药物,该药物属于代谢物组,可抑制酪氨酸分解代谢的第二阶段:将对羟基苯丙酮酸转化为尿黑酸。然而,使用这种药物会导致酪氨酸蓄积,并可能引起严重的副作用,包括角膜混浊和畏光、鼻出血和胃出血、肝功能衰竭、血液变化等。尽管如此,在美国,尼替西农已获得FDA批准用于治疗I型酪氨酸血症。[17 ],[ 18 ]
因此,针对尿黑酸尿症引起的关节问题,可以进行物理治疗,即通过运动疗法来增加肌肉力量和改善关节活动性,通过浴疗和骨质疏松疗法来减轻疼痛。
虽然酪氨酸不仅由食物提供,而且可以在体内产生,但建议患有尿黑酸尿症的患者遵循低蛋白饮食,并限制富含酪氨酸的食物的摄入,主要是牛肉和猪肉、乳制品(尤其是奶酪)、豆类、坚果和种子。
預防
基因突变无法预防,但为了防止生下有高风险先天性疾病的儿童,可以进行医学遗传咨询;对于家族史中有遗传性疾病的夫妇,在计划怀孕前,有必要进行医学遗传咨询。[ 19 ]
預測
黑黑尿症导致死亡的病例非常罕见,死亡可能由严重的心脏和肾脏并发症引起。因此,黑黑尿症患者的总体预期寿命较高。
但由于关节或脊柱剧烈疼痛,活动能力严重受限,生活质量下降,且往往呈进行性下降。