
所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。
痴呆症
該文的醫學專家

最近審查:04.07.2025

 [
[ 原因 羊角风白痴
尽管已经收集了大量有关该疾病的数据,但科学家目前仍在继续寻找有关黑蒙性白痴的病因、发病机制甚至表现的许多问题的答案。
有迹象表明该病具有遗传性。遗传类型为常染色体隐性遗传。大多数情况下,大脑半球的小脑和枕叶会受到影响,并给全身带来严重后果和并发症:视神经萎缩,神经纤维膜脱落,神经细胞之间的连接断裂。
大多数专家承认,该疾病的临床症状可能多种多样,并且与患者开始出现黑内障白痴症状的年龄有关。
在研究该病的病因时,人们注意到一种模式:该病通常影响同一家庭的子女,因此被称为“家族性黑内障白痴”。根据该病研究初期发表的研究结果,在64例黑内障白痴病例中,有37例发生在13个家庭中(每个家庭有2-5名患病儿童)。值得注意的是,在这些家庭中,患者的兄弟姐妹都非常健康。如今,人们认为隐性遗传因素在该病的发病中起着重要作用。因此,这可以解释该病在同一家庭中发病的频率。在分析遗传因素作为黑内障病因时,不应局限于患者家属是否存在临床表现(包括上行和侧线),还应考虑一些基本症状,例如视觉器官功能的特征性偏差(家族性脉络膜炎、色素性视网膜营养不良等)。
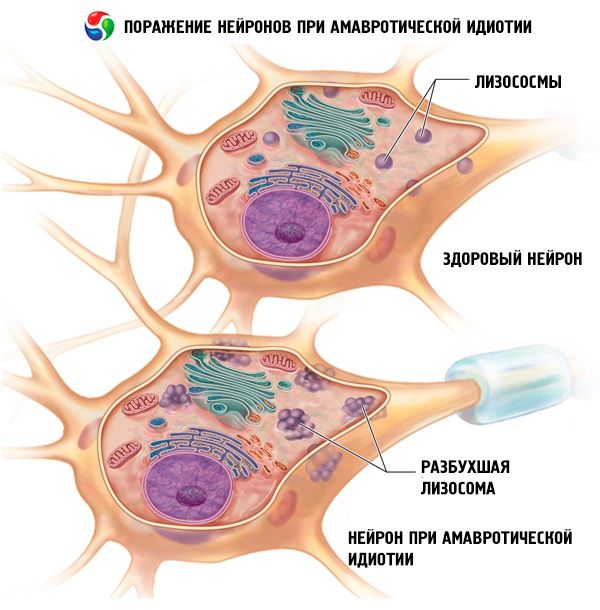
階段
婴儿型黑内障发病于4-6个月大婴儿。此类黑内障特发病具有家族性。视力迅速下降:婴儿无法凝视,无法观察物体。眼底出现所谓的“樱桃核”——黄斑区的一个红色斑点,周围有灰白色边缘。随后视神经萎缩,患儿完全丧失视力。定向力、防御性反射以及活动能力逐渐丧失。患者对声音刺激反应强烈——即使对健康人来说很轻的声音,他们也会畏缩,并可能因肌张力增高而出现抽搐。在疾病的晚期,患者会出现全身萎缩、身体虚弱以及所有伸肌张力增高。该病的预后也令人失望:患者通常在发病一年半至两年后死亡。
晚期儿童型始于3-4岁。病情进展与缓解期交替出现。智力逐渐下降,并伴有癫痫发作、协调障碍和锥体外系疾病。该型还以视神经萎缩为特征。患者在出现黑内障白痴症状后6-8年死亡。
青少年型在6-10岁时开始显现。斯皮尔迈耶黑内障白痴的进展较慢。眼底变化与色素性视网膜营养不良症的表现同时出现。患者的视力和智力逐渐下降。运动功能受损可能以不同的方式表现出来,并且不稳定:手臂和腿部瘫痪并不十分明显,锥体外系和延髓功能障碍也会出现。该疾病在出现首发症状后10-25年内会导致患者死亡。
晚期病例罕见,进展极其缓慢。患者会出现精神状态改变(类似器质性精神综合征)、视神经萎缩和视网膜色素变性。最终阶段的特征是瘫痪和癫痫样综合征。患者通常在发病后10-15年死亡。
鑑別診斷
黑内障的鉴别诊断基于特定的临床表现和眼底的特征性病理。
早期症状类似于兰丁病(一种黏多糖贮积症)。兰丁病在出生后数月内发病,并于2-3年后死亡。1/5的病例会出现眼底“樱桃核”征,视网膜退行性病变和听觉扭曲(叩诊亢进)并非其特征,但可观察到脾脏和肝脏同时肿大、精神障碍和运动障碍。
青少年型有时与 Lawrence-Moon-Biedl 综合征的表现重叠。为了区分这些疾病,需要注意它们的其他表现。Lawrence-Moon-Biedl 综合征的特征包括体重快速增加、肢体变形(以多指或多趾为特征)、明显的植物营养障碍以及运动功能障碍的缺失。
晚期黑内障白痴症状多样,使其在生前诊断变得复杂。其表现与弗里德赖希共济失调、多发性硬化症、阿尔茨海默病、皮克病、进行性瘫痪,甚至精神分裂症相似。
一些作者坚持认为,只有在患者死亡后,通过对神经系统组织学异常的分析才能可靠地确定这种疾病的诊断,尤其是在临床表现模糊的情况下。
治療 羊角风白痴
目前尚无合理有效的治疗方法。目前,黑内障白质病的治疗仅以缓解症状为目的,使用镇静剂、益智药、抗惊厥药和滋补药。
为了激活大脑的血液循环和代谢过程,医生开出了甘氨酸、埃尔卡、脑活素、谷氨酸和潘托甘。
为了缓解抽搐症状,医生开出了二苯胺或卡马西平。
使用组织提取物、输血或血浆可以获得积极的结果。