
所有iLive內容都經過醫學審查或事實檢查,以確保盡可能多的事實準確性。
我們有嚴格的採購指南,只鏈接到信譽良好的媒體網站,學術研究機構,並儘可能與醫學同行評審的研究相關聯。 請注意括號中的數字([1],[2]等)是這些研究的可點擊鏈接。
如果您認為我們的任何內容不准確,已過時或有疑問,請選擇它並按Ctrl + Enter。
特雷撤-科林斯综合征
該文的醫學專家

最近審查:04.07.2025

胎儿时期骨骼发育障碍会导致严重的颅面畸形,其中一种病症是特雷彻·柯林斯综合征 (TCS) 或下颌筋膜,即颌面骨发育不良。
根据 ICD 10 的疾病代码:XVII 类(先天性异常、变形和染色体异常),Q75.4 - 下颌面骨发育不全。
原因 特雷撤-科林斯综合征
该综合征以杰出的英国眼科医生爱德华·特雷彻·柯林斯(Edward Treacher Collins)的名字命名,他在一百多年前描述了该病症的主要特征。然而,欧洲医生更常将这种面部和颌骨异常称为弗朗切谢蒂病或综合征——这基于瑞士眼科医生阿道夫·弗朗切谢蒂(Adolf Franceschetti)的广泛研究,他在上世纪中叶提出了“下颌筋膜发育不良”这一术语。医学界也使用弗朗切谢蒂-柯林斯综合征这一名称。
特雷彻·柯林斯综合征是由TCOF1基因(位于5q31.3-33.3染色体位点)突变引起的,该基因编码一种核仁磷蛋白,负责人类胚胎颅面部分的形成。由于该蛋白的过早减少,核糖体RNA(rRNA)的生物合成和功能受到干扰。根据人类基因组研究项目的遗传学家的研究,这些过程导致神经嵴(神经沟沿线的脊状结构,在胚胎发育过程中闭合形成神经管)胚胎细胞增殖减少。
面部组织的形成源于神经嵴上部(头部)细胞的转化和分化,这些细胞沿着神经管迁移至胚胎的第一和第二鳃弓区域。这些细胞的缺乏会导致颅面畸形。畸形发生的关键时期是受精后18至28天。神经嵴细胞迁移完成后(妊娠第四周),面部几乎所有疏松间充质组织均已形成,这些疏松间充质组织随后(妊娠5至8周)分化为面部、颈部、喉部、耳部(包括内耳)以及未来牙齿各部位的骨骼和结缔组织。
發病
特雷彻·柯林斯综合征的发病机制通常具有家族性,该异常以常染色体显性遗传方式遗传,尽管也存在常染色体隐性遗传的情况(伴有其他基因突变,特别是 POLR1C 和 POLR1D)。颌面骨发育不良最难以预测的是,只有 40%-48% 的病例会遗传给孩子。也就是说,52%-60% 的患者中,特雷彻·柯林斯综合征的病因与家族异常无关,人们认为该病理是由偶发的基因新生突变引起的。最有可能的是,新的突变是妊娠期间对胎儿致畸作用的结果。
专家指出,导致该综合征的致畸因素包括大剂量乙醇、辐射、香烟烟雾、巨细胞病毒和弓形虫,以及草甘膦类除草剂(Roundal、Glyfor、Tornado 等)。医源性因素包括含13-顺式维甲酸的痤疮和脂溢性皮炎药物(异维甲酸、Accutane);抗惊厥药物苯妥英(Dilantin、Epanutin);精神药物安定、安定、利拉尼、赛达克森。
症狀 特雷撤-科林斯综合征
下颌筋膜发育不良的临床症状及其严重程度通常取决于基因突变的表现特征。在大多数情况下,这种异常的最初迹象在患儿出生后即可显现:特雷彻·柯林斯综合征患者的面部外观具有特征性。此外,形态异常通常是双侧对称的。
特雷彻·柯林斯综合征最明显的症状是:
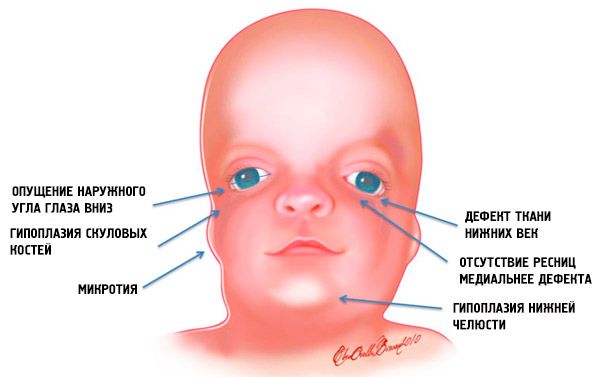
- 颅骨面部骨骼发育不全(发育不全):颧骨、额骨颧突、侧翼板、鼻旁窦、下颌骨和骨骺(髁)突出;
- 下颌骨发育不全(小颌畸形)且下颌角比正常情况更钝;
- 鼻子大小正常,但由于眉弓发育不全、颞区颧弓发育不全或缺失,显得鼻子很大;
- 眼缝向下,即眼型异常,外眼角向下垂;
- 下眼睑缺损(眼睑缺损)和部分睫毛缺失;
- 耳廓形状不规则,偏差范围很广,包括位于下颌角、无耳垂、耳屏与嘴角之间有盲瘘等;
- 外耳道狭窄或闭合(闭锁)和中耳听小骨异常;
- 腮腺唾液腺缺失或发育不全;
- 咽部发育不全(咽部和气道狭窄);
- 硬腭不融合(腭裂),以及软腭缺失、缩短或不动。
所有此类解剖异常均会引起并发症。这些并发症包括传导性听力损失或完全失聪等功能性听力障碍;由于眼球发育不良导致的视力障碍;以及腭部缺陷导致的进食和吞咽困难。此外,还存在与颌骨缺陷相关的牙齿咬合异常(错颌畸形),这会导致咀嚼和发音问题。软腭病变可以解释鼻音。
診斷 特雷撤-科林斯综合征
特雷彻·柯林斯综合征的产后诊断主要基于临床体征。当该综合征表现完整时,颅面骨发育不良很容易识别;但当病理症状表现轻微时,可能难以做出正确的诊断。
在这种情况下,应特别注意评估与异常相关的所有功能,尤其是影响呼吸的功能(因为存在睡眠呼吸暂停的风险)。还应评估和监测喂养效果和血红蛋白氧饱和度。
之后,在出生后第5-6天,需要通过听力测试来确定听力受损的程度,这应该在妇产医院进行。
需要进行检查,通过颅面畸形的荧光透视检查、全景摄影(面部颅骨结构的全景 X 射线)、各种投影的全颅计算机断层扫描、脑部 CT 或 MRI 检查进行仪器诊断,以确定内听道的状态。
如果家族史中存在特雷彻·柯林斯综合征,则最早的产前颌面畸形诊断可以通过在妊娠 10-11 周时进行绒毛膜绒毛活检来进行(该操作可能导致流产和子宫感染)。
还对家庭成员进行血液检查;在怀孕 16-17 周时,进行羊水分析(经腹羊膜穿刺术);在怀孕 18-20 周时,进行胎儿镜检查并从胎盘的胎儿血管中抽取血液。
但最常见的是,超声波用于胎儿(怀孕 20-24 周)对该综合征的产前诊断。
需要什麼測試?
治療 特雷撤-科林斯综合征
与所有遗传性先天性缺陷病例一样,严重特雷彻·柯林斯综合征的治疗也仅限于姑息治疗,因为目前尚无针对此类病症的治疗方法。该综合征的畸形范围广泛,程度也十分严重,因此,医疗干预的性质和强度也存在诸多选择。
助听器用于矫正和改善听力,言语治疗用于改善言语。
对于严重的气道狭窄(需进行气管切开术)和喉部狭窄(需进行胃造口术以便进食)的情况,需要在早期进行手术干预。有时也可能需要进行腭部矫正手术。
下颌延长手术通常在2-3岁或以后进行。软组织重建包括下眼睑缺损矫正和耳廓整形手术。
預測
这种病症的预后如何?取决于畸形程度和症状强度。特雷彻·柯林斯综合征是终身疾病。
 [ 25 ]
[ 25 ]